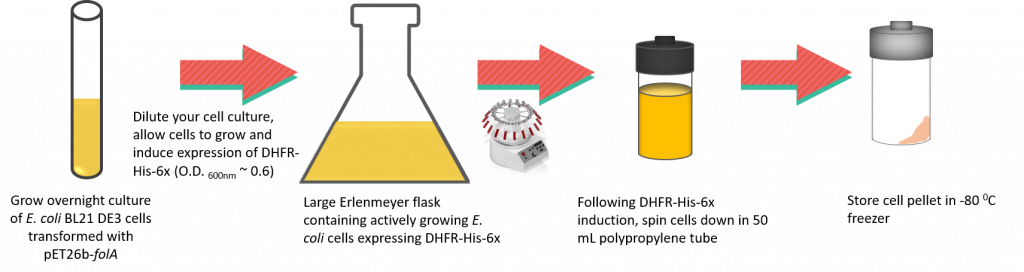
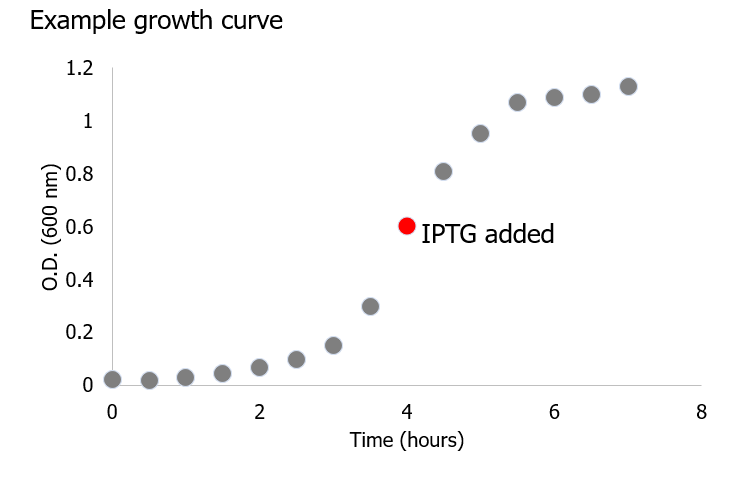
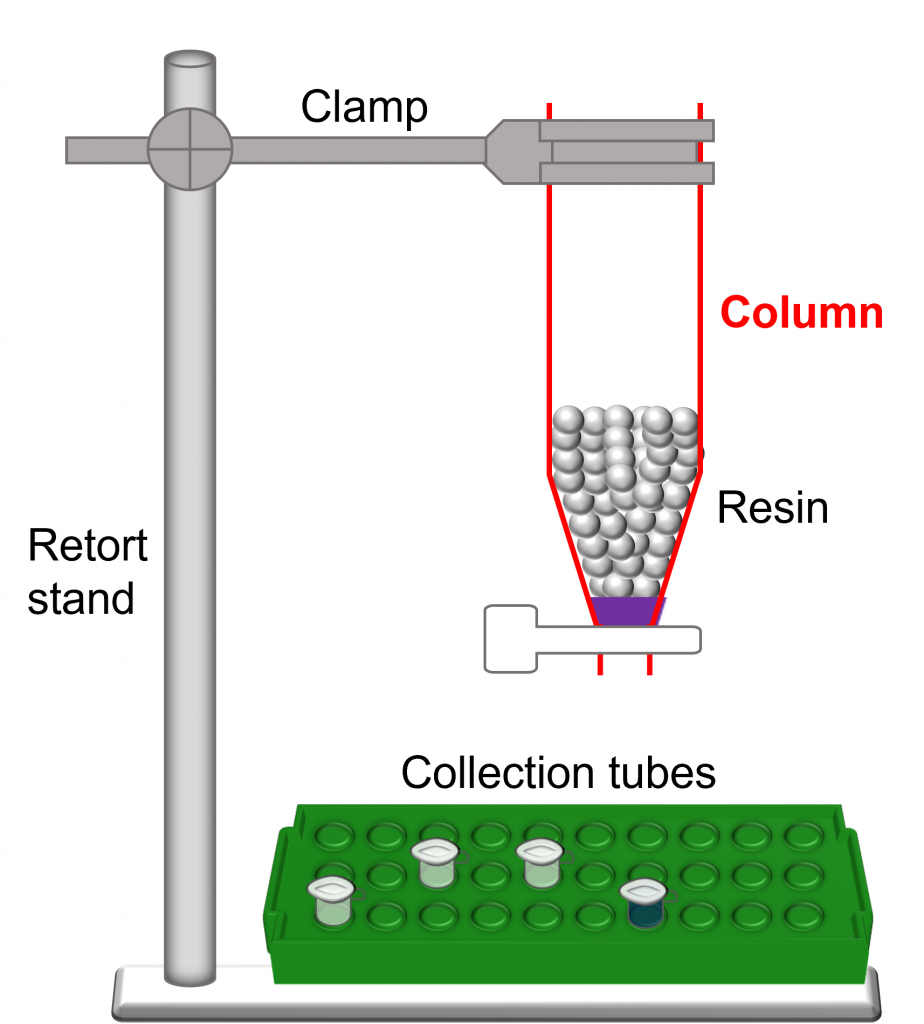
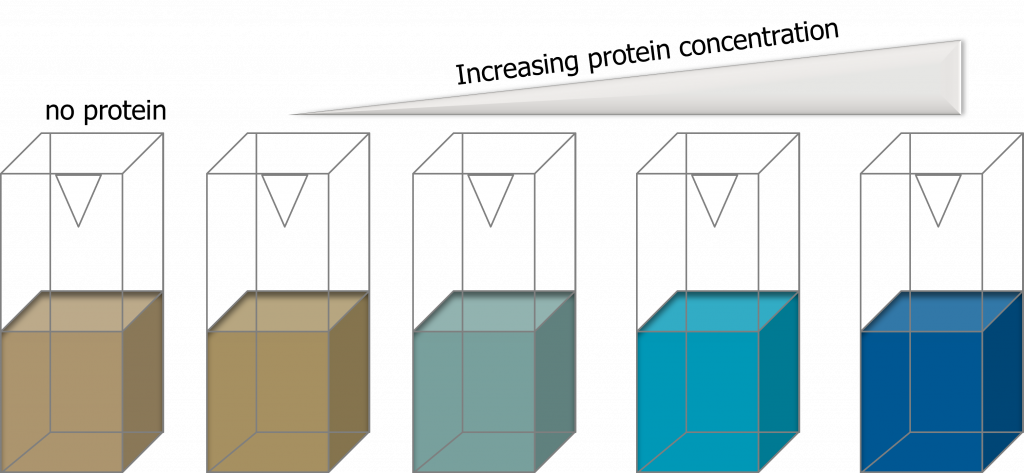
(1)Cohen, S. N.; Chang, A. C.; Boyer, H. W.; Helling, R. B. Construction of biologically functional bacterial plasmids in vitro. PNAS 1973, 70, 3240–3244.
(2) Kresge, N.; Simoni, R. D.; Hill, R. L. The Characterization of Restriction Endonucleases: the Work of Hamilton Smith J. Biol. Chem. 2010, 285, e2-e3.
(3) Kelly, S.; Kaddurah-Daouk, R.; Smith, H. O. Purification of the HhaII Restriction Endonuclease from an Overproducer Escherichia coli Clone J. Biol. Chem. 1985, 260, 15339–15344.
(4) Kaddurah-Daouk, R.; Cho, P.; Smith, H. O. Catalytic Properties of the HhaII Restriction Endonuclease J. Biol. Chem. 1985, 260, 15345–15351.
(5) Arber, W.; Linn, S. DNA modification and restriction. Ann. Rev. of Biochem. 1969, 38, 467–500.
(6) Brownlee, C. Danna and Nathans: Restriction enzymes and the boon to molecular biology. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5909.
(7) Danna, K.; Nathans, D. Specific changes of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc. Natl. Acad. Sci. U.S.A. 1971, 68, 2913–2917.
(8) Villa-Komaroff, L.; Efstratiadis, A.; Broome, S.; Lomedico, P.; Tizard, R.; Naber, S. P.; Chick, W. L.; Gilbert, W. A bacterial clone synthesizing proinsulin. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 3727–3731.
(9) Pray, L. The biotechnology revolution: PCR and the use of reverse transcriptase to clone expressed genes. Nature Education, 2008, 1, 1.
(10) Mullis, K. B.; Faloona, F. A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Meth. Enzym. 1987, 155, 335–350.
(11) DNA Technology Milestones collection. http://www.nature.com/milestones/miledna/pdf/miledna_all.pdf (accessed July 10, 2013).
(12) Profiles in Science: The Joshua Lederberg Papers. http://profiles.nlm.nih.gov/BB/ (accessed July 10, 2013).
(13) Boyer, R. Biochemistry Laboratory: Modern Theory and Techniques, 2nd ed.; Pearson Education Inc., 2012; pp. 309-324.
(14) Taylor, J. R. An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements, 2nd ed.; University Science Books, 1999 pp. 128–129.
(15) Davidson College, Department of Biology, How to use a micropipettor. http://www.bio.davidson.edu/courses/Molbio/Protocols/pipette.html (accessed May 15, 2021), © Copyright 2002 Department of Biology, Davidson College, Davidson, NC 28036.
(16) University of Regina, Department of Biology, proper use of pipetters. http://bio305lab.wikidot.com/appendix:pipettes (accessed May 15, 2021)
(17) Reed, R.; Holmes, D.; Weyers, J.; Jones, A. Practical Skills in Biomolecular Sciences, 2nd edition.; Prentice Hall, 2003.
(18) Shakhashiri, Z. Chemical Demonstrations: A Handbook for Teachers of Chemistry. Volume 2; University of Wisconsin Press, 1985.
(19) Seidman, L. A. Basic Laboratory Calculations for Biotechnology, Pearson Education Inc.; 2008.
(20) Voet, D.; Voet, J. D.; Pratt, C. Fundamentals of Biochemistry, 3rd ed.; John Wiley and sons Inc., 2008.
(21) Standard deviation by Robert Niles. https://www.robertniles.com/stats/stdev.shtml (accessed May 15, 2021).
(22) Newton, C.R.; Graham, A. PCR, 2nd edition.; BIOS Scientific Publishers, Oxford, 1997; pp. 1-27.
(23) The Nobel Prize in Chemistry 1993, Nobelprize.org. 13 Jun 2013. http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1993/ (accessed May 15, 2021)
(24) Glaser, J.A. Validity of Nucleic Acid Purities Monitored by 260 nm/280 nm Absorbance Ratios, BioTechniques, 1995, 18, 62-63.
(25) Thein, S. L.; Wallace R. B. (1986). The use of synthetic oligonucleotides as specific hybridization probes in the diagnosis of genetic disorders. In Human genetic diseases: a practical approach, Davis, K. E.), IRL Press, Herndon, Virginia, 1986; p. 33-50.
(26) Innis, M. A.; Gelfand, D. H.; Sninsky, J. J.; White, T. J. PCR protocols: a guide to methods and applications. Academic Press; 1990.
(27) Lawyer, F. C.; Stoffel, S.; Saiki, R. K.; Myambo, K.; Drummond, R.; Gelfand, D. H. 1989. Isolation, characterization, and expression in Escherichia coli of the DNA polymerase gene from Thermus aquaticus. J. Biol. Chem.1989, 264: 6427-6437.
(28) Pearson Custom Library; 2016 – “Making Decisions with Data” chapter obtained from the following book: Hage, D. S., & Carr, J. D. (2011). Analytical chemistry and quantitative analysis. Boston: Prentice Hall.
(29) Bokor, J. R., & Darwiche, H. (2011b, October). Pipetting by design. Paper presented at the annual conference of the National Association of Biology Teachers, Anaheim, CA.
Other references of interest:
a. McPherson, M. J.; Moller, S. G. PCR. BIOS Scientific Publishers Ltd., 2000; pp. 1-83.
b. Suggs, S. V.; Wallace, R. B.; Hirose, T.; Kawashima, E. H.; Itakura, K. (1983) Use of synthetic oligonucleotides as hybridization probes: isolation of cloned cDNA sequences for human beta 2-microglobulin. Proc Natl Acad Sci U S A.1983, 78, 6613-6617.
c. Eckert, K. A.; Kunkel, T. A. DNA polymerase fidelity and the polymerase chain reaction. Genome Res. 1991, 1, 17-24.
d. Kolmodin, L.A.; Williams, J.F. Polymerase Chain Reaction: Basic Principles and Routine Practice, The Nucleic Acid Protocols Handbook; Humana Press, Totowa, New Jersey, 2000; pp. 569-580.








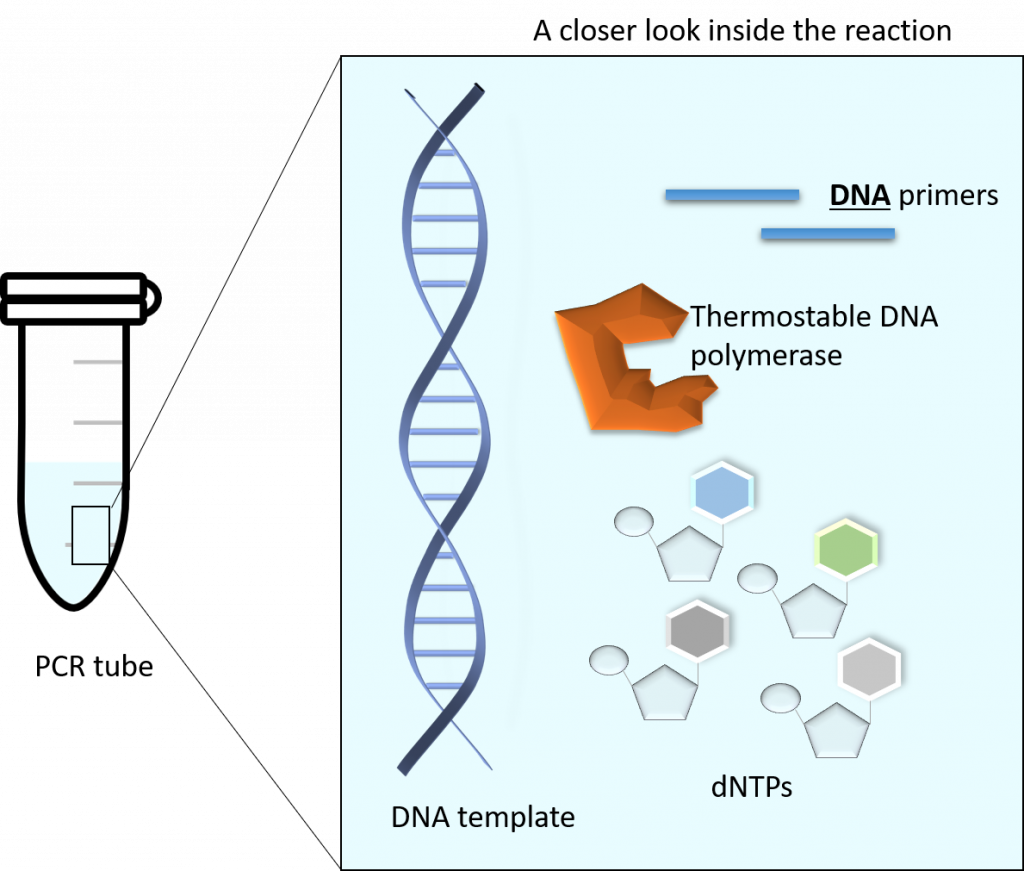
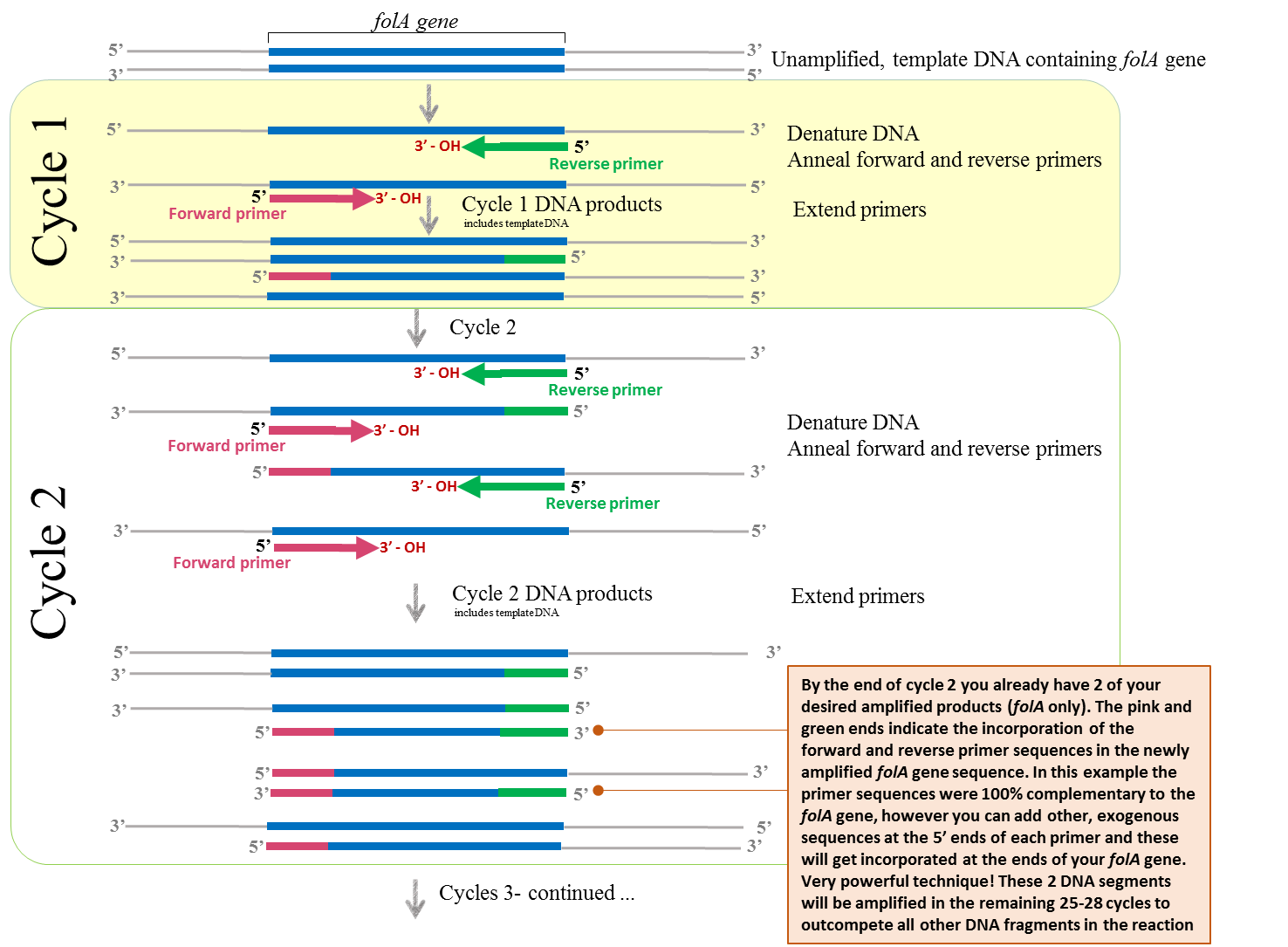
 95°C – (
95°C – (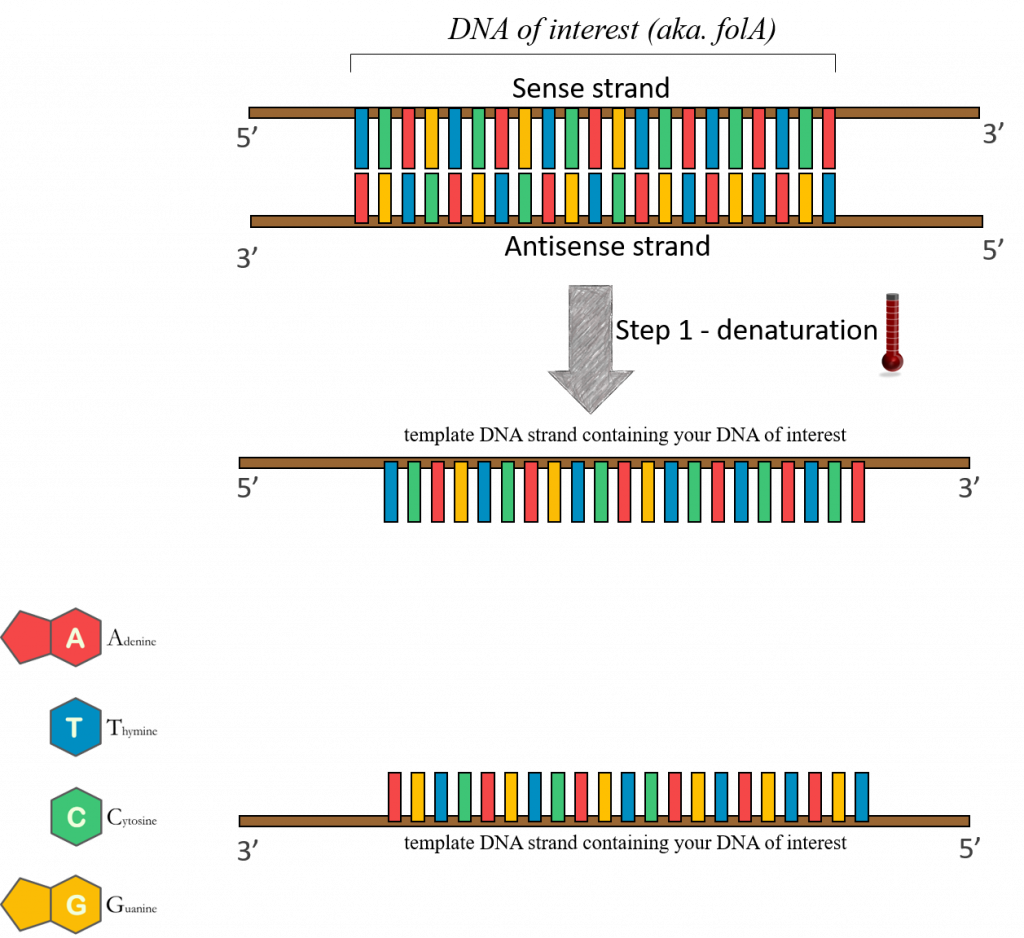
 37-72°C – (
37-72°C – (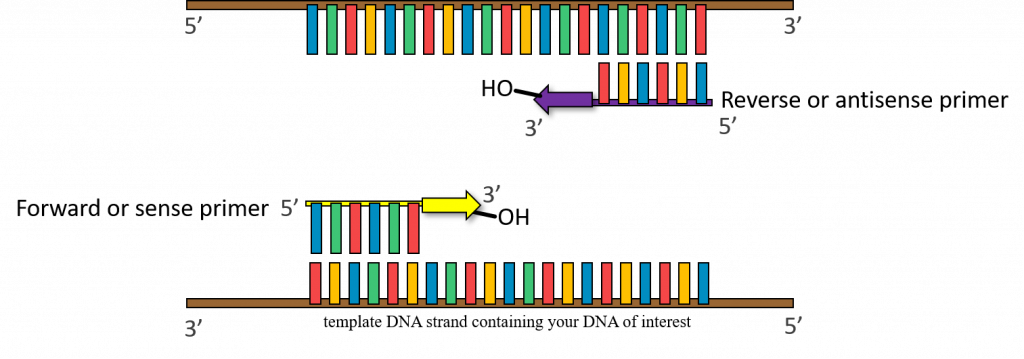
 72°C – (
72°C – (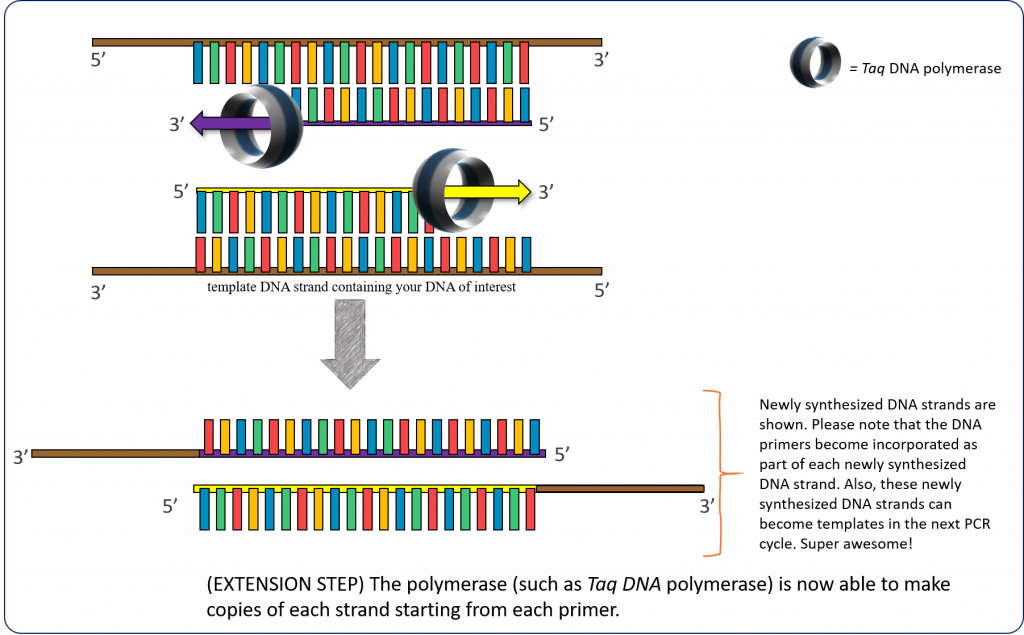



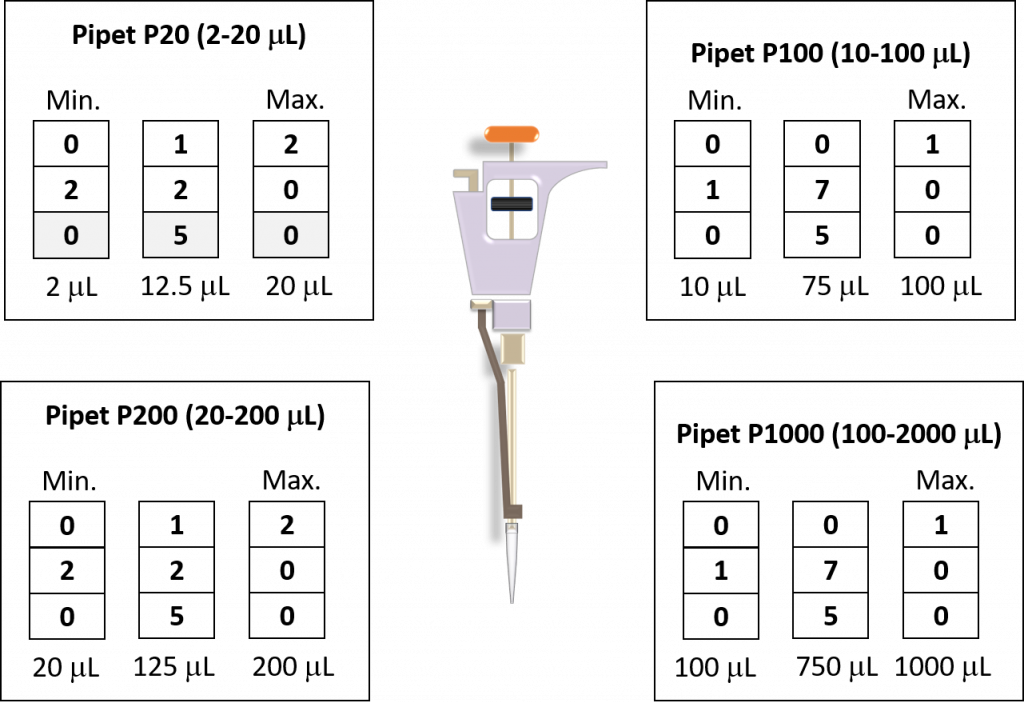
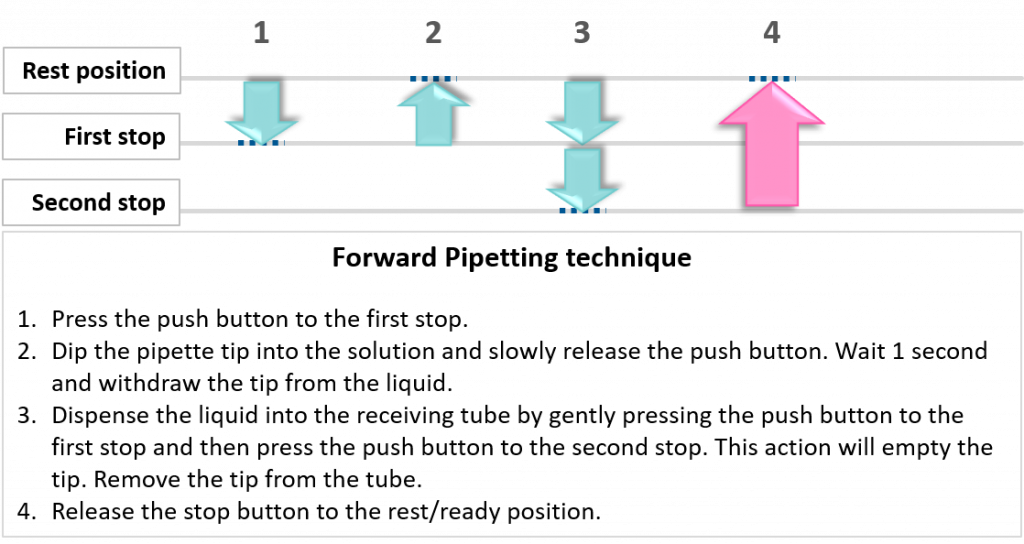


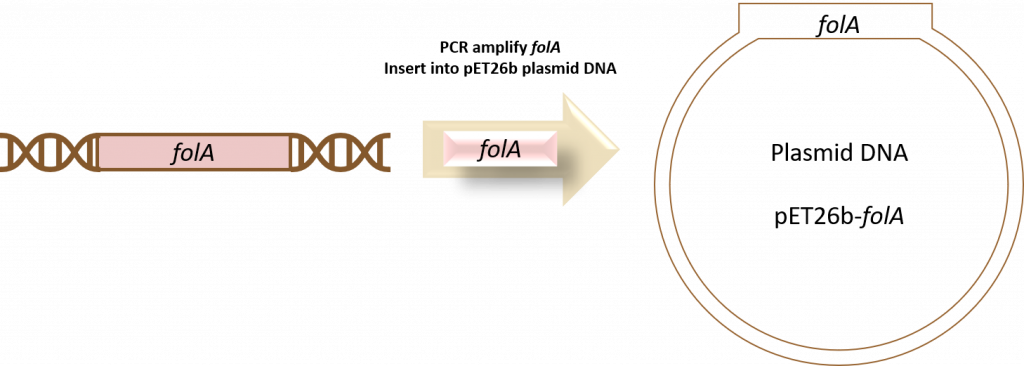
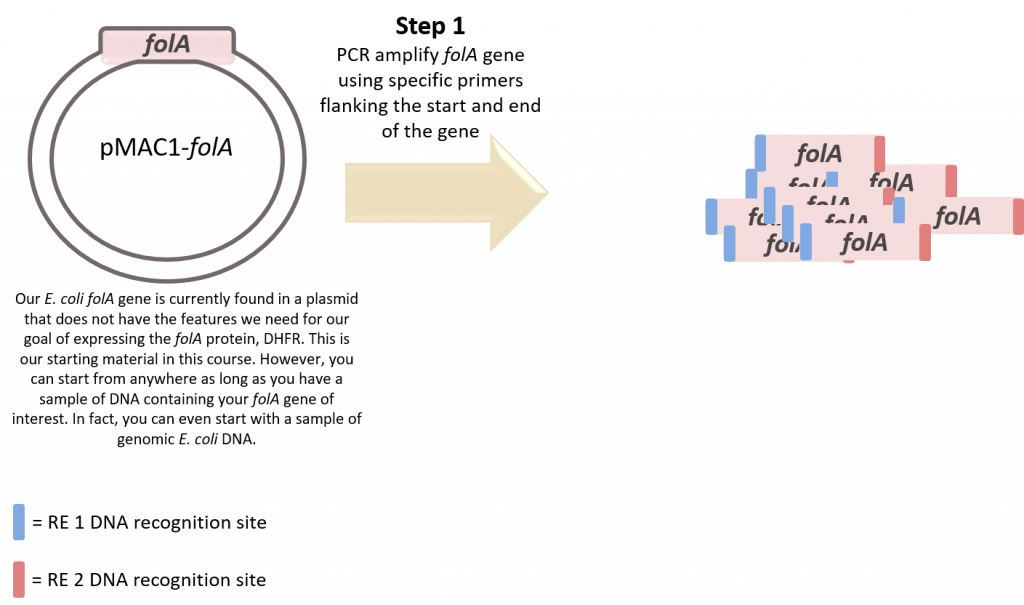
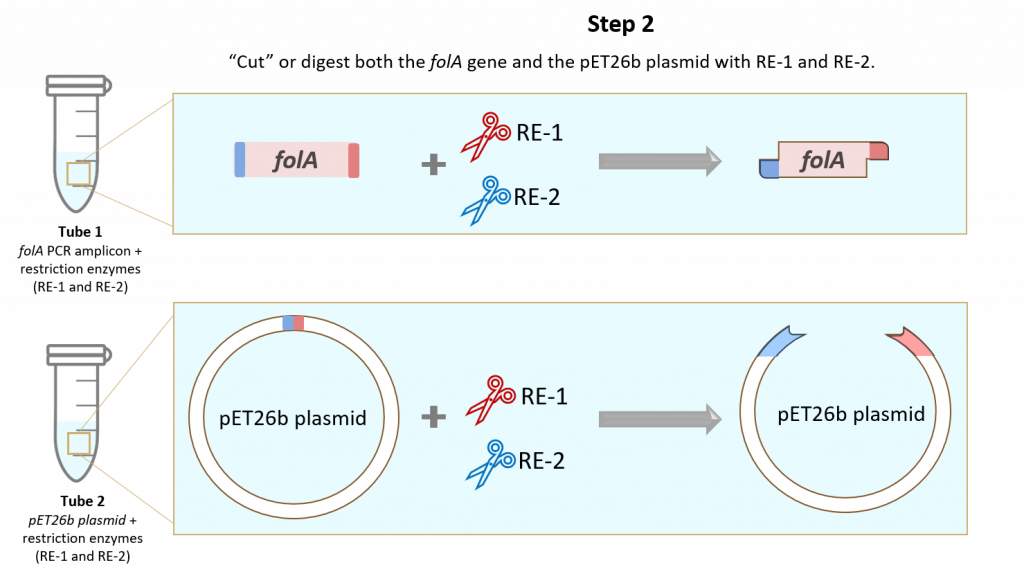
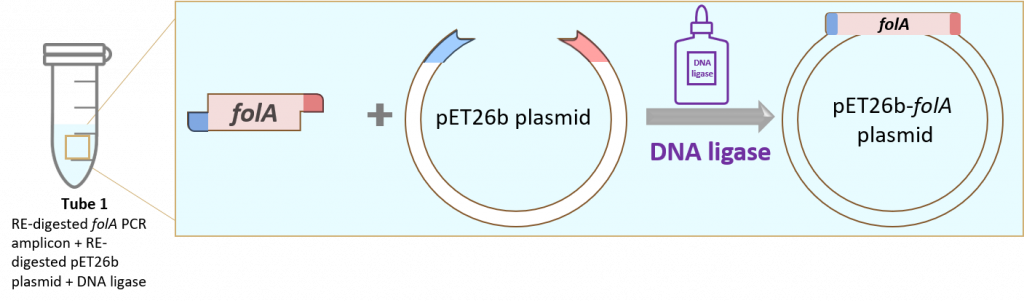


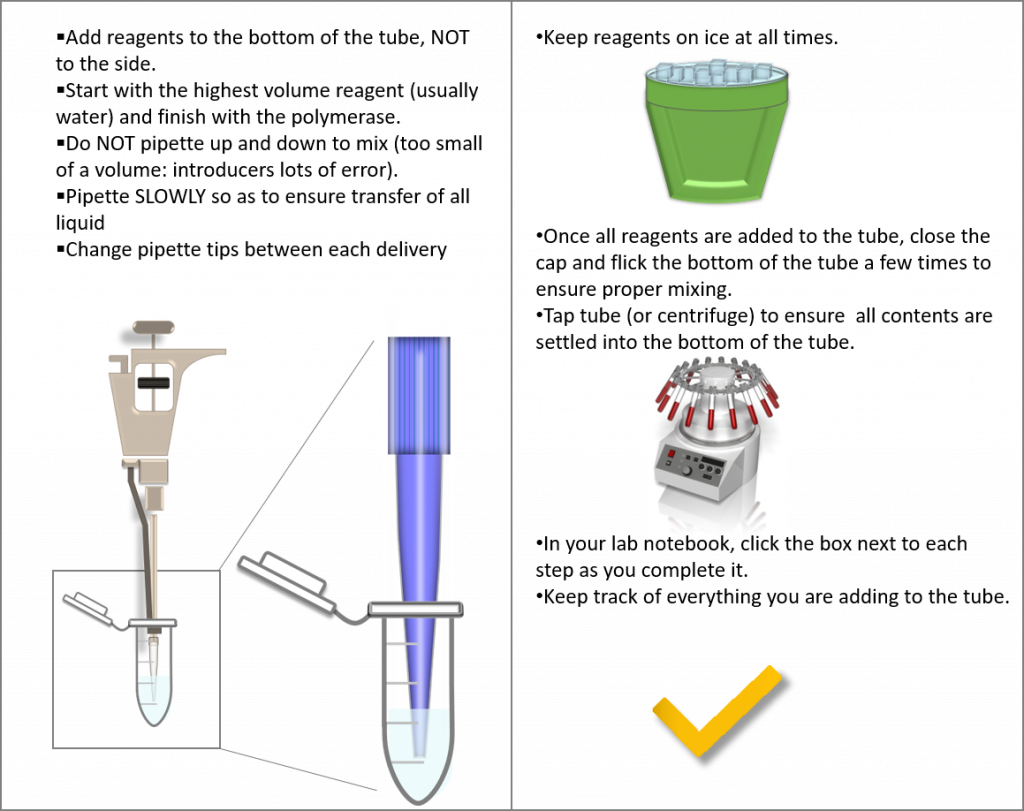
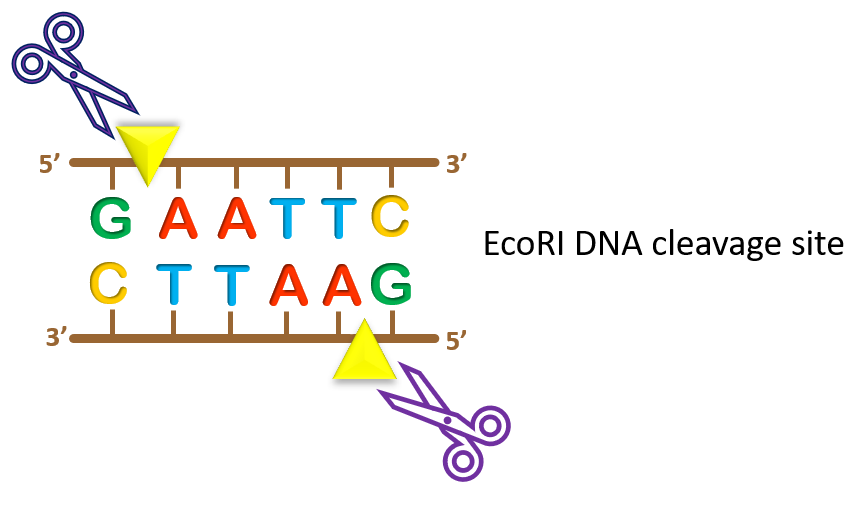

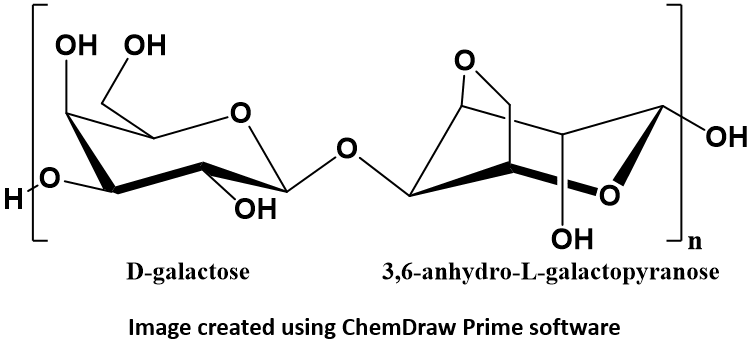
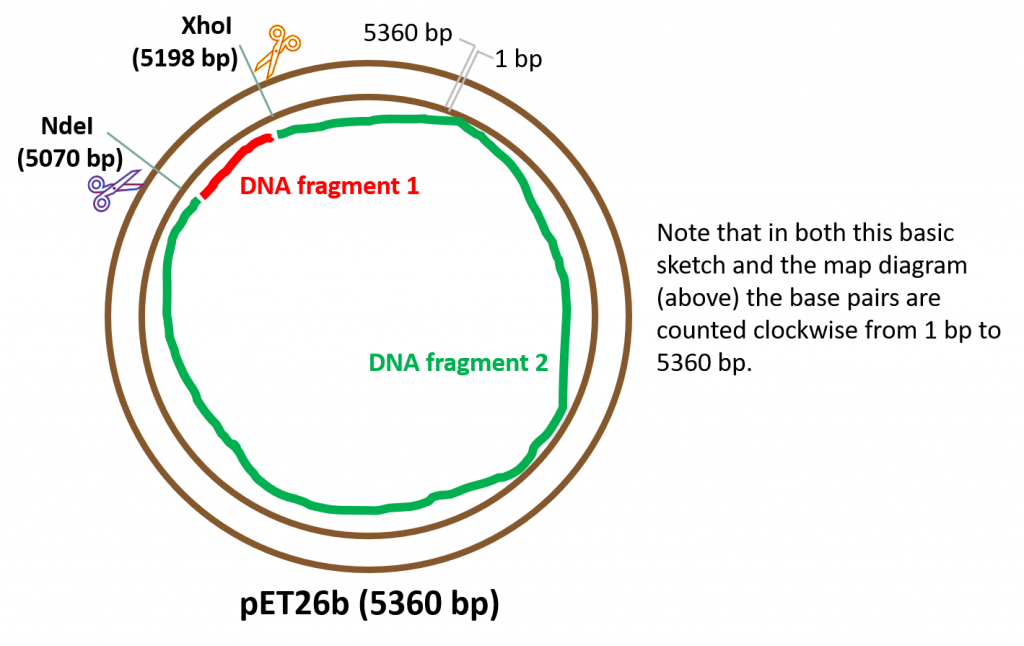
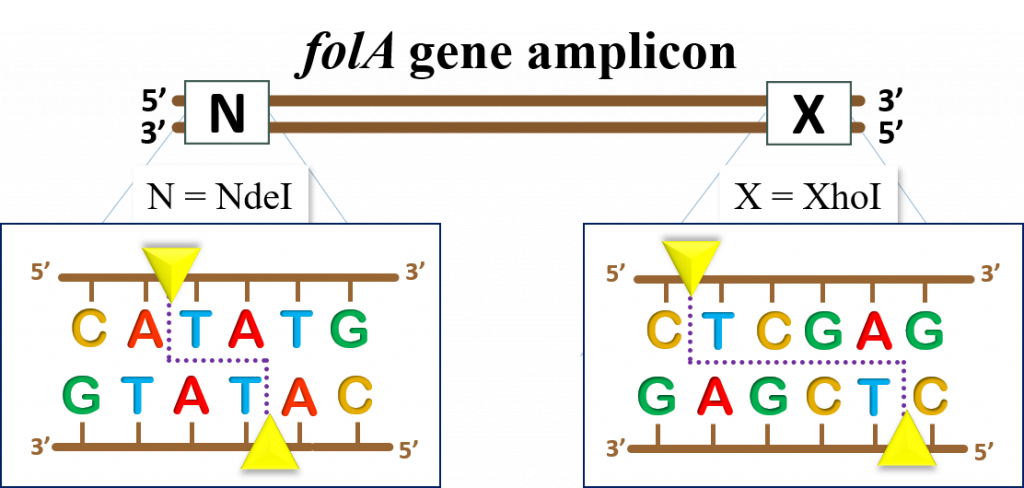
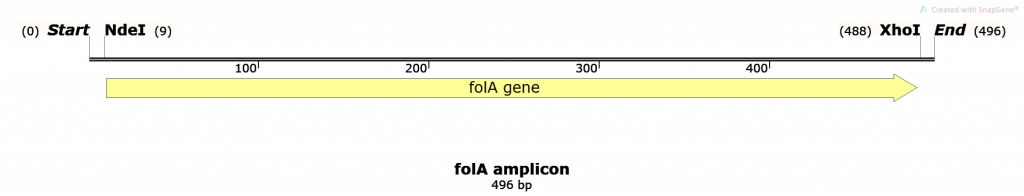

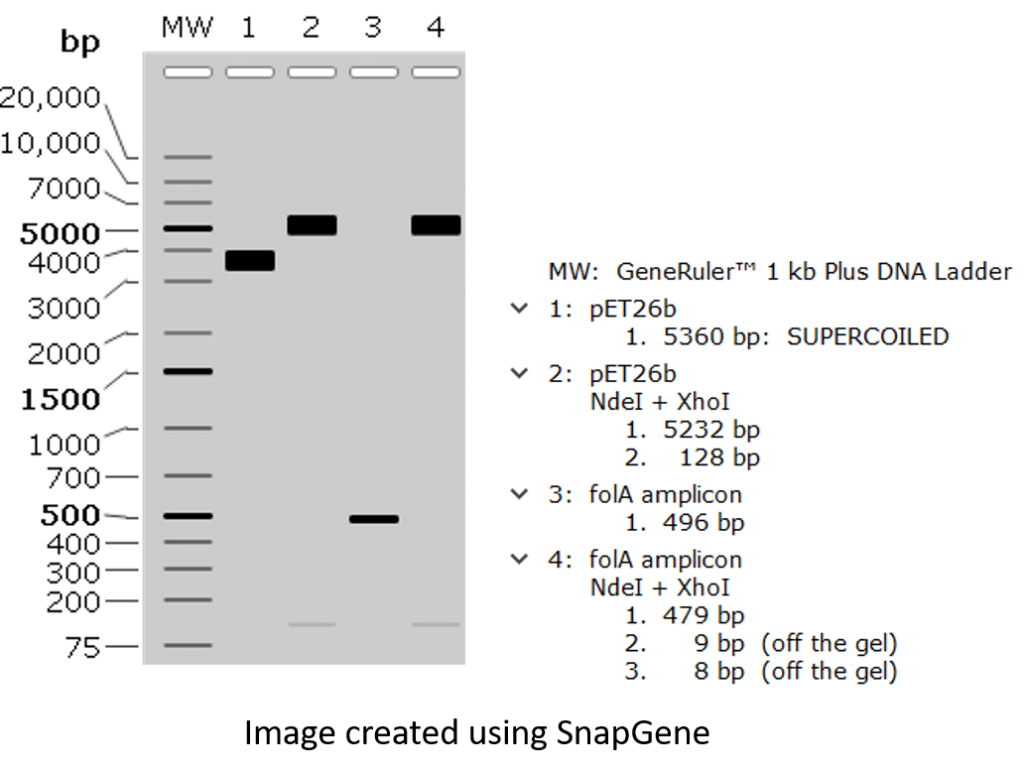
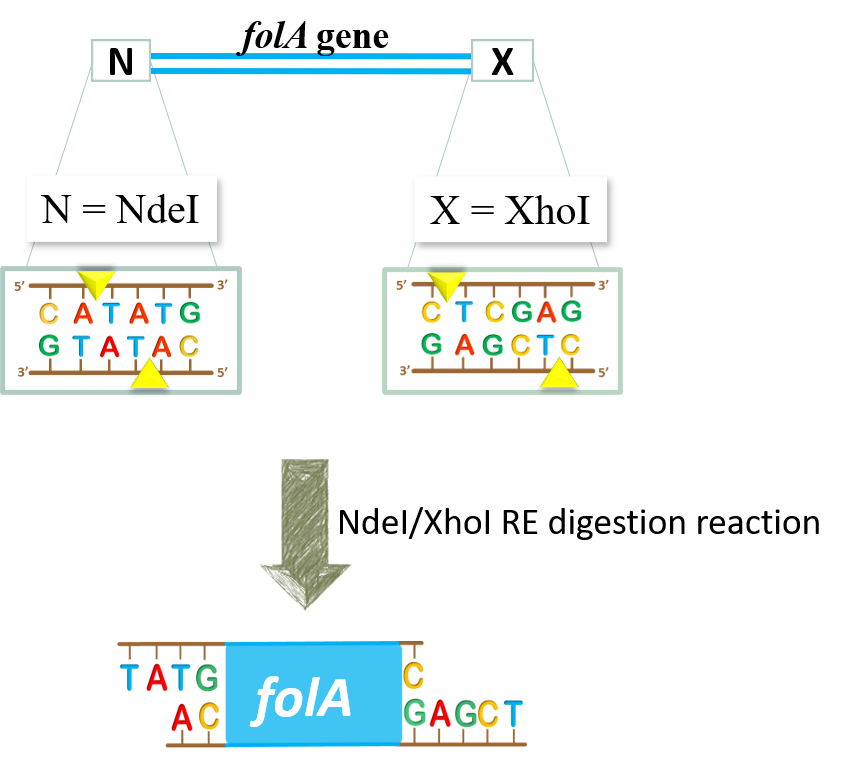


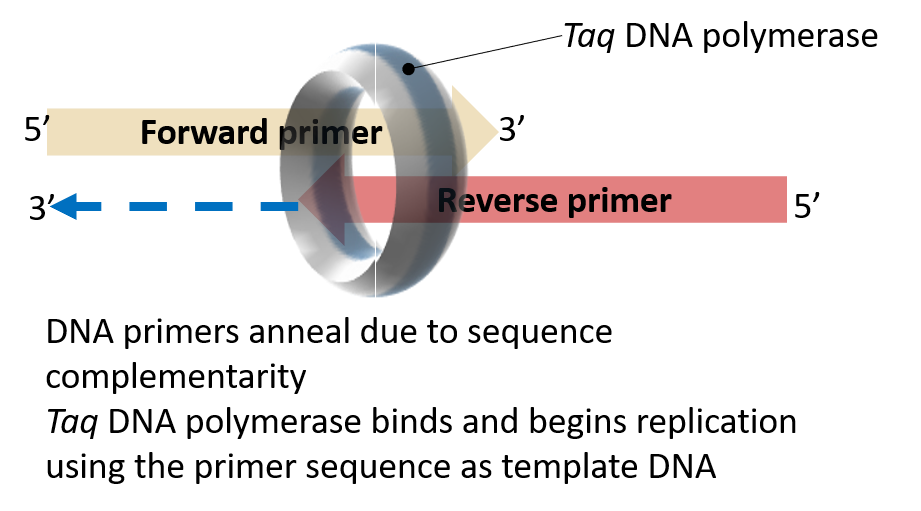
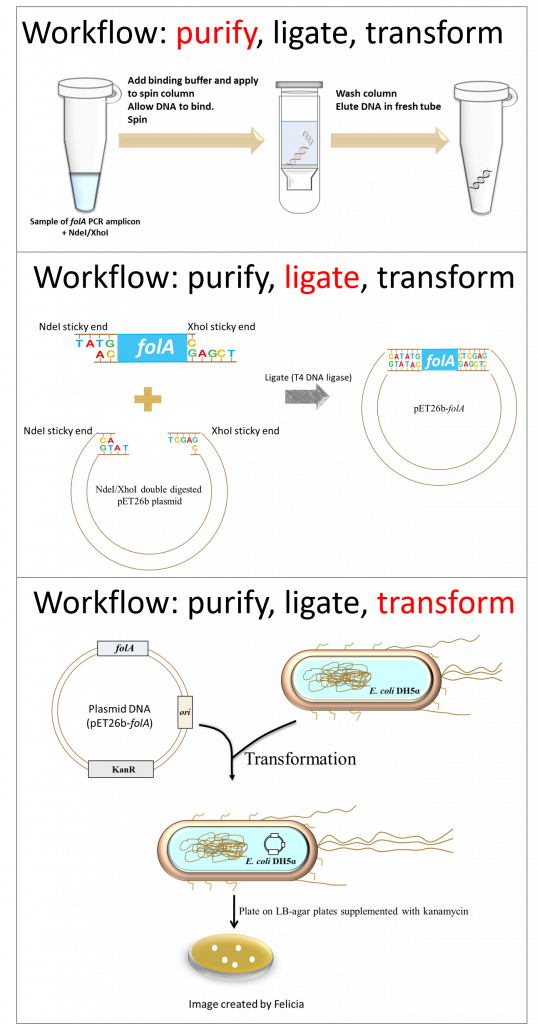

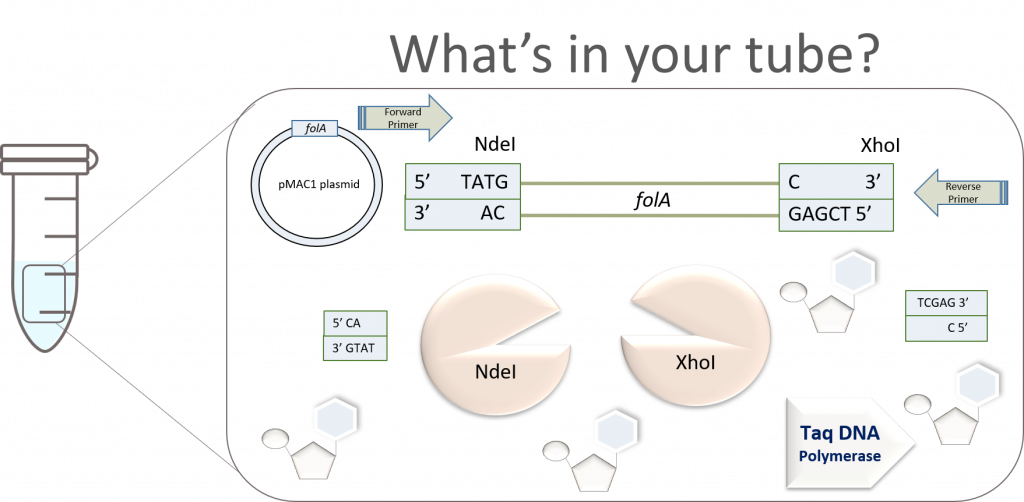
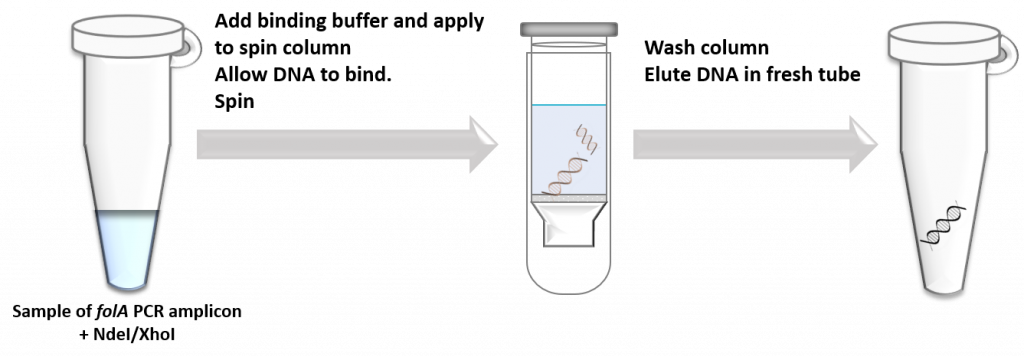
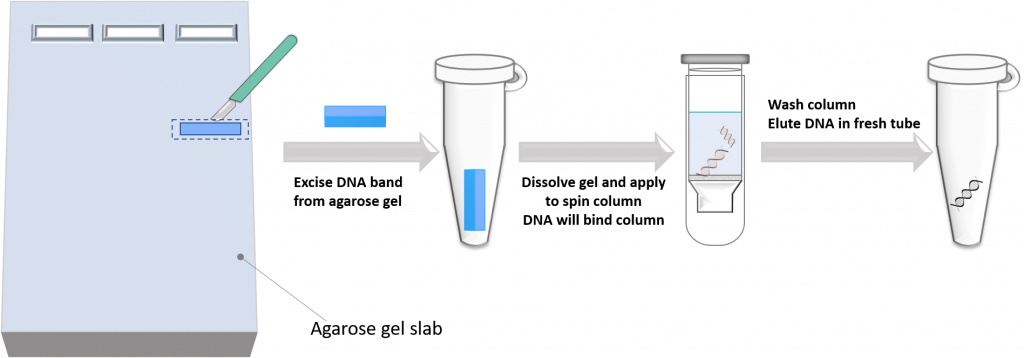
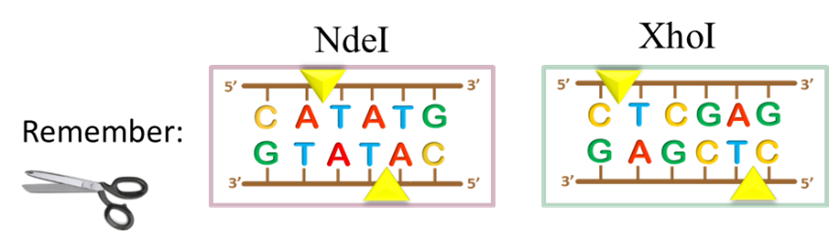
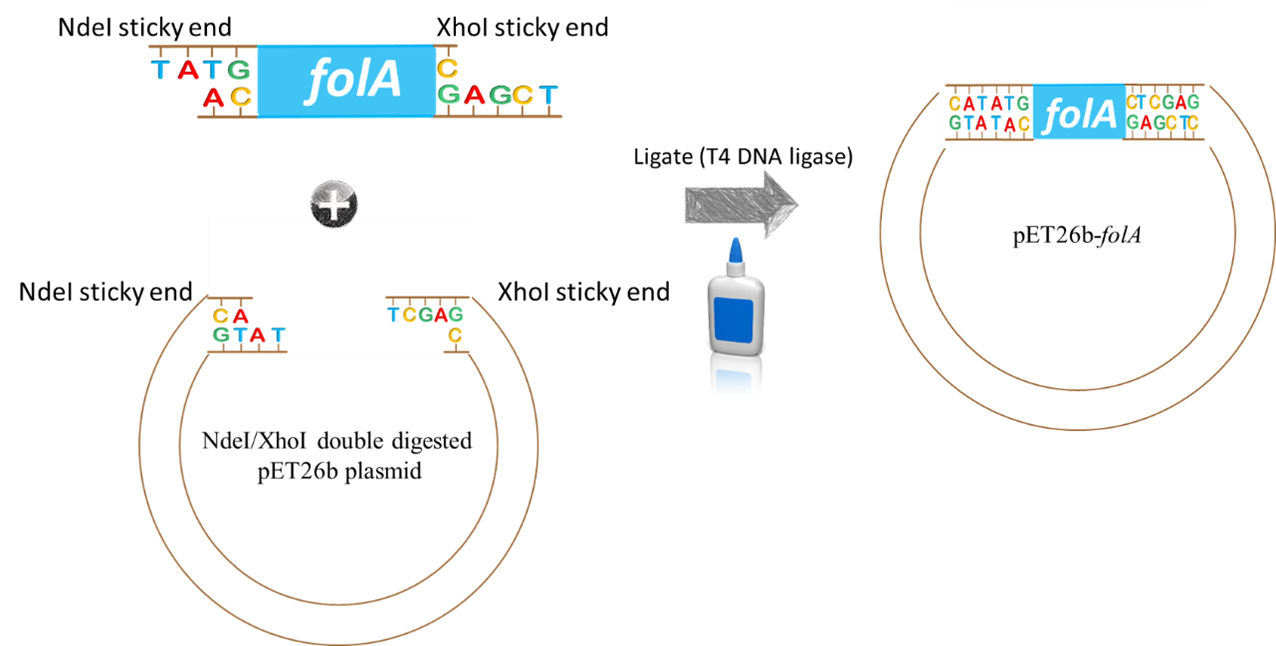
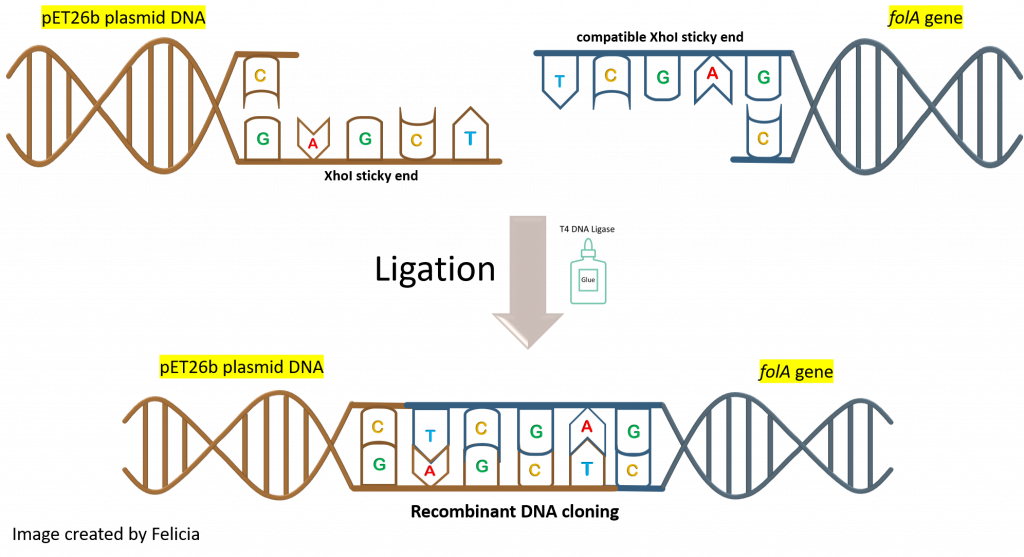
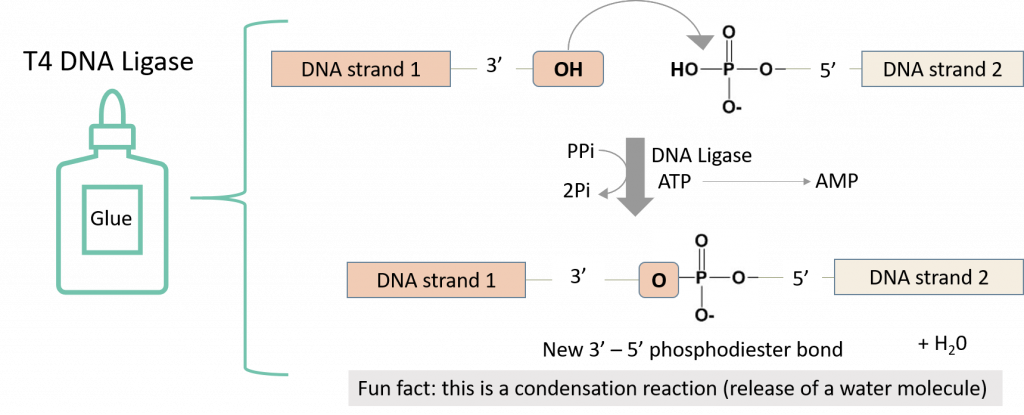
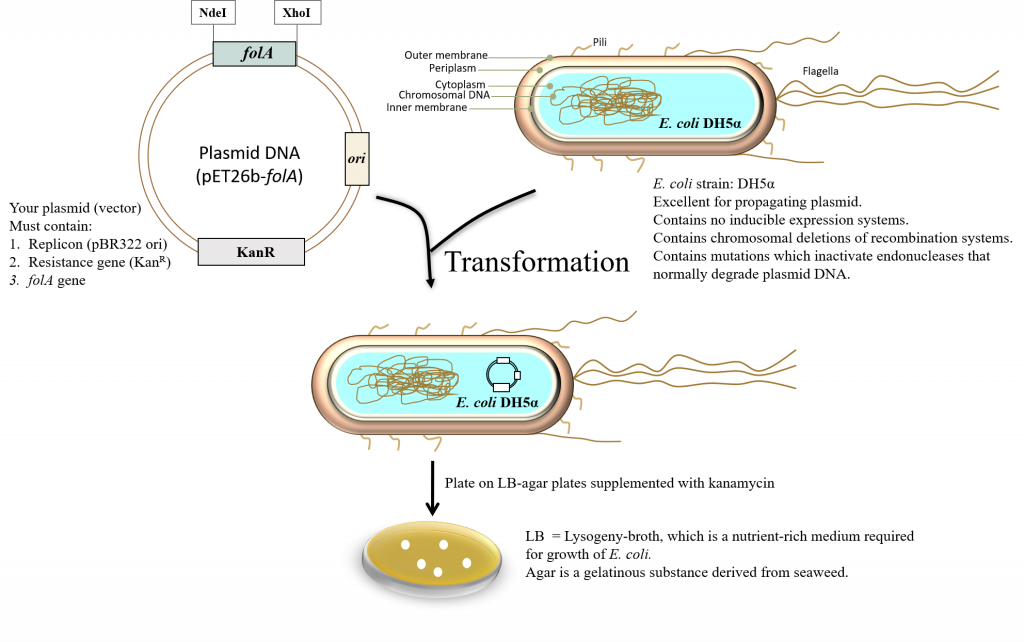
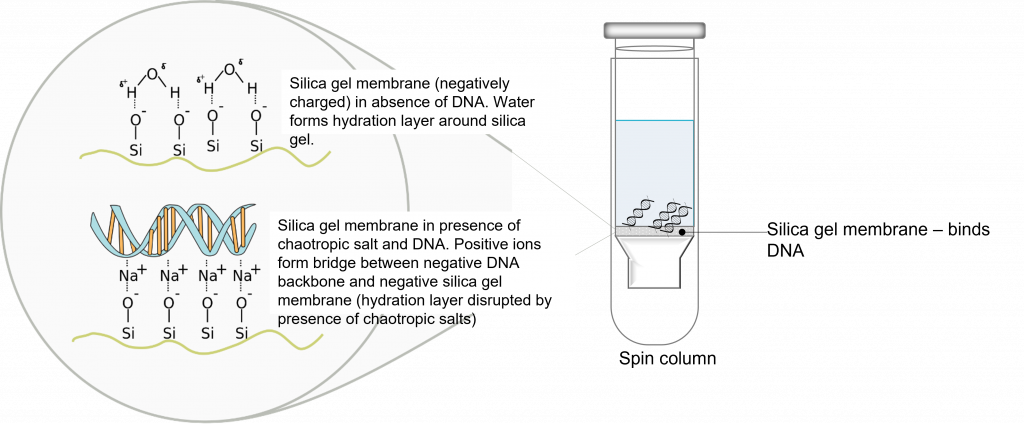


 key twice.
key twice. key.
key.